COVID-19
Searching for molecular clues
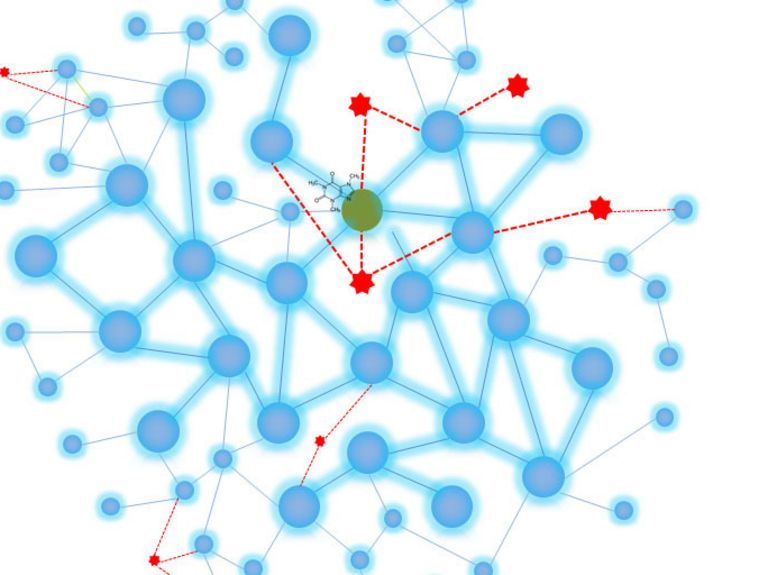
Graphic: Pascal Falter-Braun
Researchers working on an EU-funded project at the Helmholtz Zentrum Munich are analyzing whether drugs that have already been approved might be suitable for treating COVID-19. To get to the bottom of this, the researchers are trying to understand which proteins, signal paths, and molecular structures the virus exploits and modifies in the human body.
Pascal Falter-Braun from the Helmholtz Zentrum Munich, German Research Center for Environmental Health (HMGU), coordinates the EU project RiPCoN (Rapid interaction profiling of 2019-nCoV for network-based deep drug-repurpose learning). Together with partners from France and Spain, his team is looking at molecular interactions between the virus and humans.
Mr. Falter-Braun, you’re the director of the Institute of Network Biology at the HMGU. The Institute hasn’t typically focused on viruses prior to this. What’s the starting point for your project?
We’re researching how the molecular networks within a cell work. What happens there is very complex because the numerous proteins in the cell don’t work alone. Rather, they always perform their functions with other proteins as part of a network. One important function is handling signals, for instance during processes that regulate cell proliferation, which has to be accelerated at some points and slowed down at others. Immune responses are another example. These networks basically have a similar structure to social and other complex networks, which is both fascinating and significant. Some proteins have very strong networks, similar to an influencer or an important air hub—but other proteins tend to stay on the sidelines and are highly focused on independent functions.
How are you now making use of this knowledge?
All viruses penetrate human cells and multiply there. Proteins belonging to the virus—including the novel coronavirus—are then produced which, in turn, interact with the cell’s own protein network in the host, or person, and try to reprogram it. We look at this interplay at the level of the proteins so we can understand exactly what the virus is doing. We want to know which factors influence it, and that’s exactly what our project is based on.
And how do you go about this, given that you’re currently working from home yourself ...
The initial phase of work in the lab has already been underway for several weeks now despite the fact that operations are being kept to a minimum, which is challenging. We’ve had the virus’s gene segments synthesized and are now analyzing how they interact with nearly every human protein—there are around 18,000 of them. And we are doing this for all human coronaviruses, so not just for SARS-CoV-2, but also for MERS, the SARS virus that emerged in 2003, and four others that are less harmful. This will help us gain a more in-depth understanding of this entire class of viruses.

Pascal Falter-Braun from the Helmholtz Zentrum Munich, German Research Center for Environmental Health (HMGU), coordinates the EU project RiPCoN (Rapid interaction profiling of 2019-nCoV for network-based deep drug-repurpose learning). Image: HMGU
You’re using a yeast-based system for your analyses. How exactly does that work?
We use a special trick. To put it in very simple terms, we use genetic engineering methods that cause one viral and one human protein each to be produced in a yeast cell. These yeast cells are prepared in such a way that they only survive and divide if the human and viral proteins bind to each other. If they bond, this shows that the proteins interact with each another at the molecular level. We can then use sequencing to trace the pairs where bonding occurred out of the millions we’re testing.
What do you then do with this information?
We have an advantage because we already understand a lot about the network of protein interactions in humans and know which functions a very large number of human proteins are involved in. Network maps, which look like subway maps, are also a very good way of illustrating this. Now we want to investigate which of these processes the viral proteins interfere in, which networks are located nearby, and above all, the illnesses in which the processes attacked by the viruses play a part. We want to know how to correct the processes that are impaired by the virus so we can find a way to prevent the virus from multiplying.
Do analyses involving drugs come into play here as well then?
Yes. In the next stage of our project, we want to examine which existing drugs already influence the network at these points and could therefore be suitable for neutralizing SARS-CoV-2 as well. This would make the time-consuming approvals process unnecessary or speed it up. Drugs that are already effective against viruses have a high likelihood of being a good match in general. Many experiments elsewhere are naturally looking at this as well. Our niche is that we can identify less obvious preparations that might be suitable.
... and you use artificial intelligence and machine learning to support your work.
Based on the information generated, we can use computer models to help us find out what chances individual drugs have of being successful. We’ll take absolutely every drug that has been approved into consideration. Using AI to support our work is incredibly important given the vast volumes of data involved. Our aim is to identify the points in the network that are all affected by the virus, illnesses, and drugs. Thanks to our partners in France and Spain, we can combine all the related information. Our partners in France have also developed models that make it possible to predict interactions between proteins and RNA molecules. This is key, because coronaviruses always start by inserting an RNA molecule into the human host cell.
Would this project and its models have been possible five years ago?
No, or only parts of it, and we wouldn’t have been able to get nearly as much information from those parts.
How soon do you think you’ll have results?
The network maps and the other analyses being carried out by our partners will take some time. My hope is that we’ll have the first candidates in three to four months so we can then conduct in vitro experiments with them.
And do you feel any pressure in terms of other people’s expectations, especially considering how urgent the situation is?
We rather tend to put pressure on ourselves, because you naturally feel a sense of responsibility when you get approval to do a project like this. At the same time, the prospect of doing something that makes a difference in this situation is extremely motivating. And we’ll definitely be able to contribute to the general understanding of coronaviruses—even if we don’t “deliver a drug,” although we are optimistic that this will be the case.
There are plans for a second phase of the project as well. You want to find out the effects that naturally occurring genetic differences in the interacting human and viral proteins have on how the disease progresses in individual cases. Can you explain the idea behind this?
Everyone has slightly different genetic information for many of our proteins. Among other things, this means that some of us are more prone to certain diseases—from allergies, to cancer, to hereditary diseases. We can almost certainly assume that it’s also easier for viral proteins to interact with some people’s proteins, according to how they are formed. Depending on where this “enhanced interaction” occurs, this could lead to the disease progressing in different ways: It might be more severe, or it might be milder as well—for example, in the case of proteins that are more capable of intercepting the virus. In other words, the key question is: What proteins does the virus target, and what variations in these proteins do humans have? In the subsequent analysis, we then want to combine our answers with real data on the disease and draw conclusions for other patients on this basis. We’d like to start doing this as quickly as possible, too. It will take a lot of work, and we’ll need to use data from other research projects. But I’m confident that it will be worth it because the insights we’ll gain can be used for risk management and resource planning at hospitals in the future.
Thank you for the interview!






Readers comments